sobereva 发表于 2020-1-2 18:34
1 是
2 是。对于一般意义的SAPT,考察哪两个的作用,就把哪两个结构拿出来当做一个二聚体做SAPT。

sobereva 发表于 2020-1-2 18:34
1 是
2 是。对于一般意义的SAPT,考察哪两个的作用,就把哪两个结构拿出来当做一个二聚体做SAPT。
captain 发表于 2020-1-2 20:18
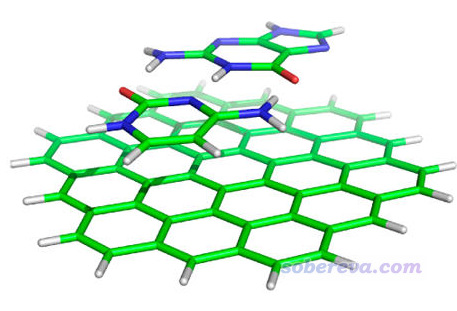
有感于楼主的问题,我也想请问大神
比如一个A-B-C三聚体体系,计算A-C之间的作用,直接把A-C提取出来当 ...
hzfish 发表于 2020-1-3 10:02
可能会有明显的多体效应。
下面是GKS-EDA的计算结果,可以参考。
captain 发表于 2020-1-3 10:21
十分感谢!
请问这篇文献出自哪里?能否给个链接?
hzfish 发表于 2020-1-3 10:39
The Journal of Chemical Physics 131, 014102 (2009); doi: 10.1063/1.3159673

hzfish 发表于 2020-1-3 10:02
可能会有明显的多体效应。
下面是GKS-EDA的计算结果,可以参考。
Peter_zhong 发表于 2020-1-3 11:41
老师 我有点疑惑,1.这个多体效应数值是可以接受(忽略的)的范围吗,还是算大? 2.还有做GKS-EDA计算一 ...
wuzhiyi 发表于 2020-1-4 05:18
想问一下老师,研究三聚体下,两个分子之间的相互作用,fispat这方法如何?
| 欢迎光临 计算化学公社 (http://bbs.keinsci.com/) | Powered by Discuz! X3.3 |