GoldenBaby 发表于 2021-10-25 11:08
你好像没有尝试计算精确的Hessian矩阵吧
Q-Chem employs a Switching/Gaussian or “SWIG” implementation of these PCMs.523, 524, 525, 373, 521 This approach resolves a long-standing—though little-publicized—problem with standard PCMs, namely, that the boundary-element methods used to discretize the solute/continuum interface may lead to discontinuities in the potential energy surface for the solute molecule. These discontinuities inhibit convergence of geometry optimizations, introduce serious artifacts in vibrational frequency calculations, and make ab initio molecular dynamics calculations virtually impossible.523, 524 In contrast, Q-Chem’s SWIG PCMs afford potential energy surfaces that are rigorously continuous and smooth. Unlike earlier attempts to obtain smooth PCMs, the SWIG approach largely preserves the properties of the underlying integral-equation solvent models, so that solvation energies and molecular surface areas are hardly affected by the smoothing procedure.
scf 发表于 2021-10-25 04:45
qchem里有一种SWIG方法,据说收敛会好
https://manual.q-chem.com/5.2/Ch12.S2.html
sobereva 发表于 2021-10-25 11:33
上传一堆输出文件的时候,明确说清楚每个文件各对应什么,别让答疑者一个个去打开查看、去猜,不仅耽误答疑 ...
scf 发表于 2021-10-25 11:45
qchem里有一种SWIG方法,据说收敛会好
https://manual.q-chem.com/5.2/Ch12.S2.html
红米饭1234 发表于 2021-10-25 09:23
谢谢sob老师,我在仔细看看,因为源参考文献里面的溶剂是MTBE,想着甲苯和这个相似,因为做的Pd催化有机反 ...
wzkchem5 发表于 2021-10-25 16:47
我有点怀疑MTBE和甲苯能不能看作是足够相似的。假如能的话,那就意味着MTBE的极性也非常低,所以opt freq ...
biogon 发表于 2021-10-25 10:09
MTBE是醚,介电常数目测和甲苯差了很大了
wzkchem5 发表于 2021-10-25 16:47
我有点怀疑MTBE和甲苯能不能看作是足够相似的。假如能的话,那就意味着MTBE的极性也非常低,所以opt freq ...
biogon 发表于 2021-10-25 17:09
MTBE是醚,介电常数目测和甲苯差了很大了
wzkchem5 发表于 2021-10-25 16:47
我有点怀疑MTBE和甲苯能不能看作是足够相似的。假如能的话,那就意味着MTBE的极性也非常低,所以opt freq ...
红米饭1234 发表于 2021-10-27 02:58
MTBE的介电常数和甲苯差不多呀
sai77 发表于 2021-10-29 09:00
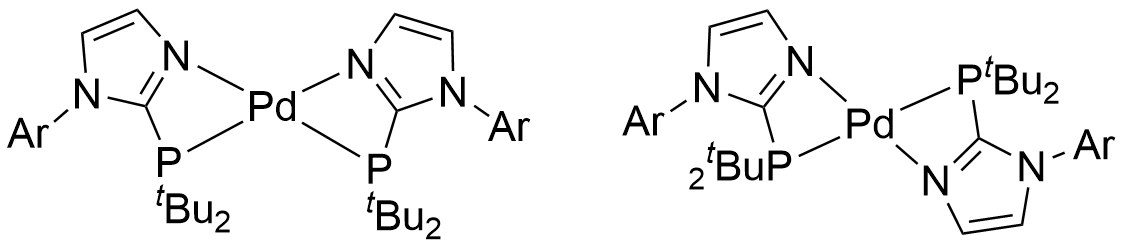
我怎么感觉你这初始结构不太合理,P上两个叔丁基这么大位阻,反式配位能量更低些吧
红米饭1234 发表于 2021-10-31 10:13
啥叫反式配位啊
红米饭1234 发表于 2021-10-31 17:13
啥叫反式配位啊
| 欢迎光临 计算化学公社 (http://bbs.keinsci.com/) | Powered by Discuz! X3.3 |