本帖配套链接:
程序主页 https://www.maplechem.org/
Github:https://github.com/ClickFF/MAPLE
DOI: https://doi.org/10.1039/D6SC01279E
| 任务类型 | #task(method=...) 关键字 |
| 几何优化 #opt | lbfgs(默认)、rfo、sd、cg、sdcg |
| 过渡态搜索 #ts | neb(可附 refine=cineb/refine=nebts)、prfo、dimer、string(可附 refine=stringts)、autoneb |
| 反应路径 #irc | 积分器 gs / lqa / hpc / eulerpc |
| 振动分析 #freq | mw(默认,质量加权)/ nonmw / both;RRHO 热化学量含 Grimme/Truhlar/Minenkov 低频校正 |
| 势能面扫描 #scan | 1D 到多维;底层与 #opt 同(relaxed 模式) |
| 分子动力学 #md | 系综 nve / nvt / npt;恒温器 Langevin/Bussi/Nosé-Hoover;恒压 c-rescale (目前还在完善中,PBC的支持还在更新中) |
| MLIP 模型 #model | ani1x / ani2x / ani1ccx / ani1xnr / aimnet2 / aimnet2nse / maceoff23s/m/l / maceomol / macepols/m/l / egret / uma |
| 额外功能 | D4 色散校正(仅 ANI 系列)、GBSA 隐式溶剂、周期性边界(#pbc)(还在完善中)、轨迹重启、DCD 输出 |
ΔE_rxn= E(P) − E(R) = (−234.595946 − (−234.525473)) Eh = -0.070473 × 627.5094kcal/mol= −44.2 kcal/mol
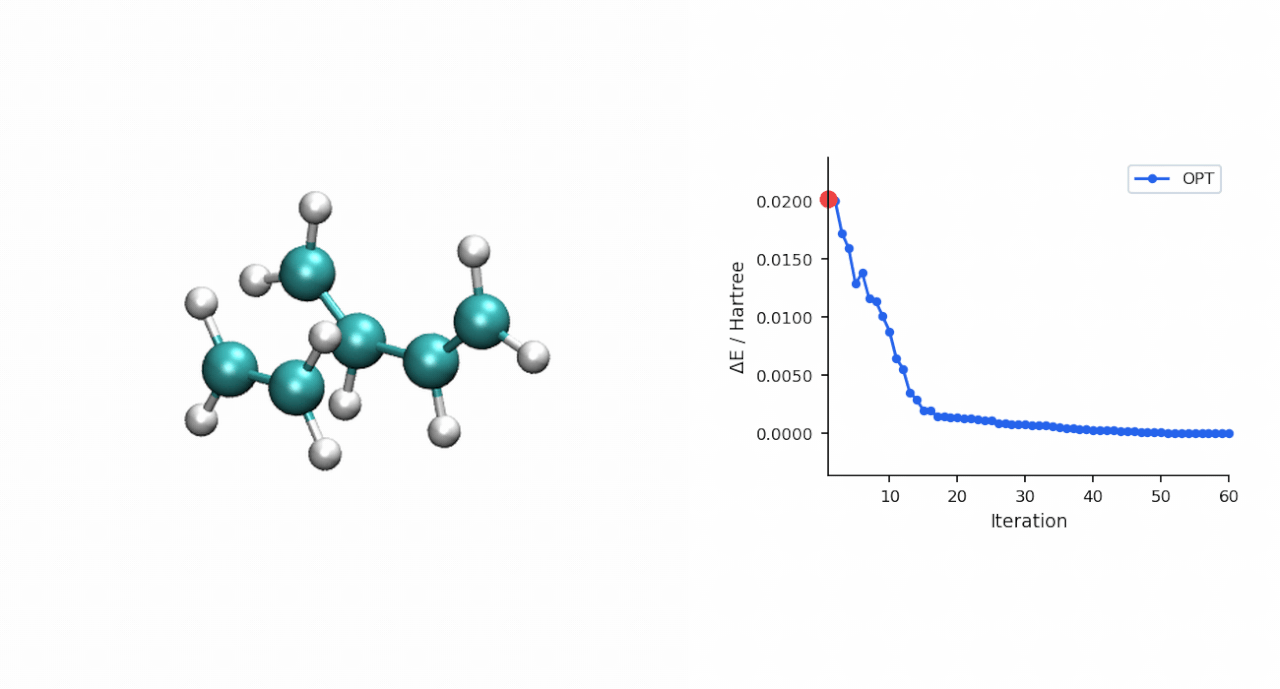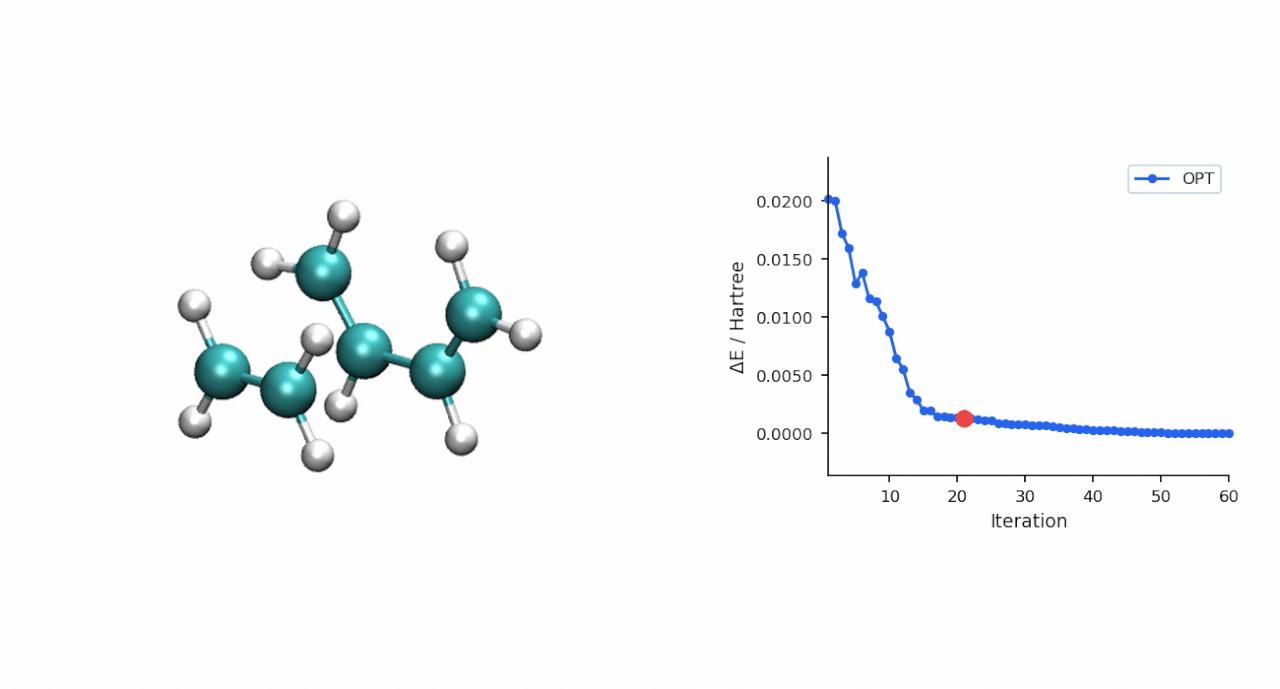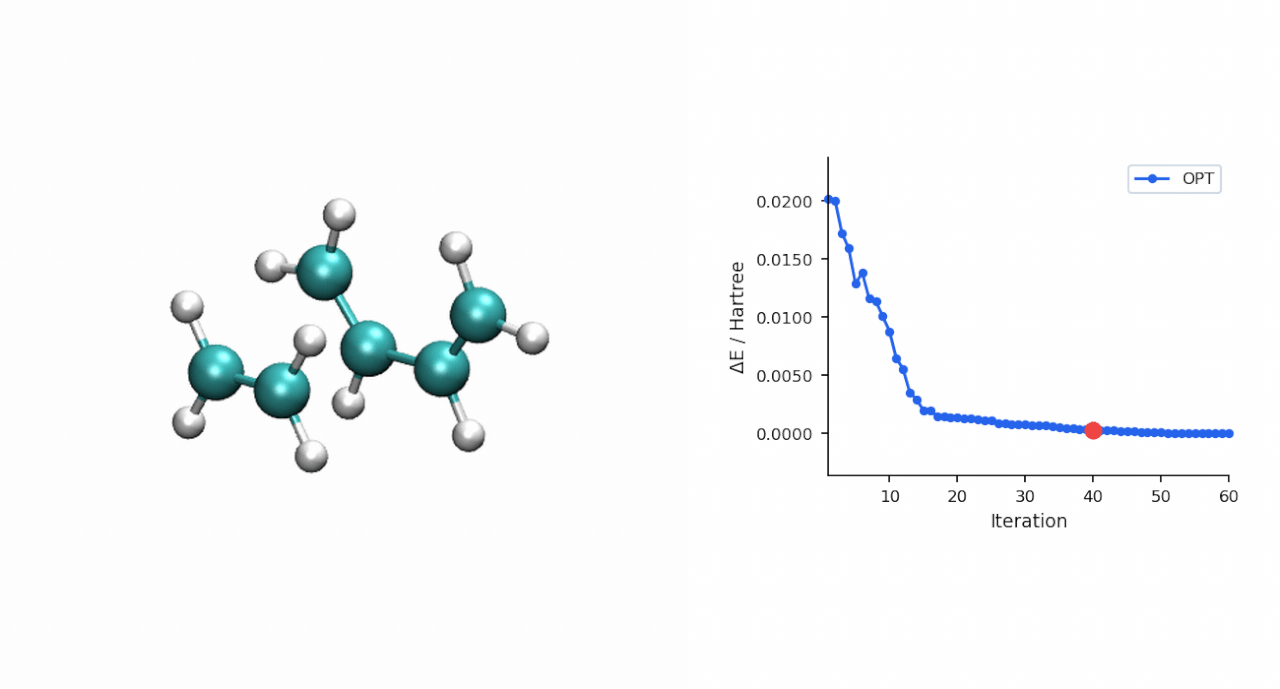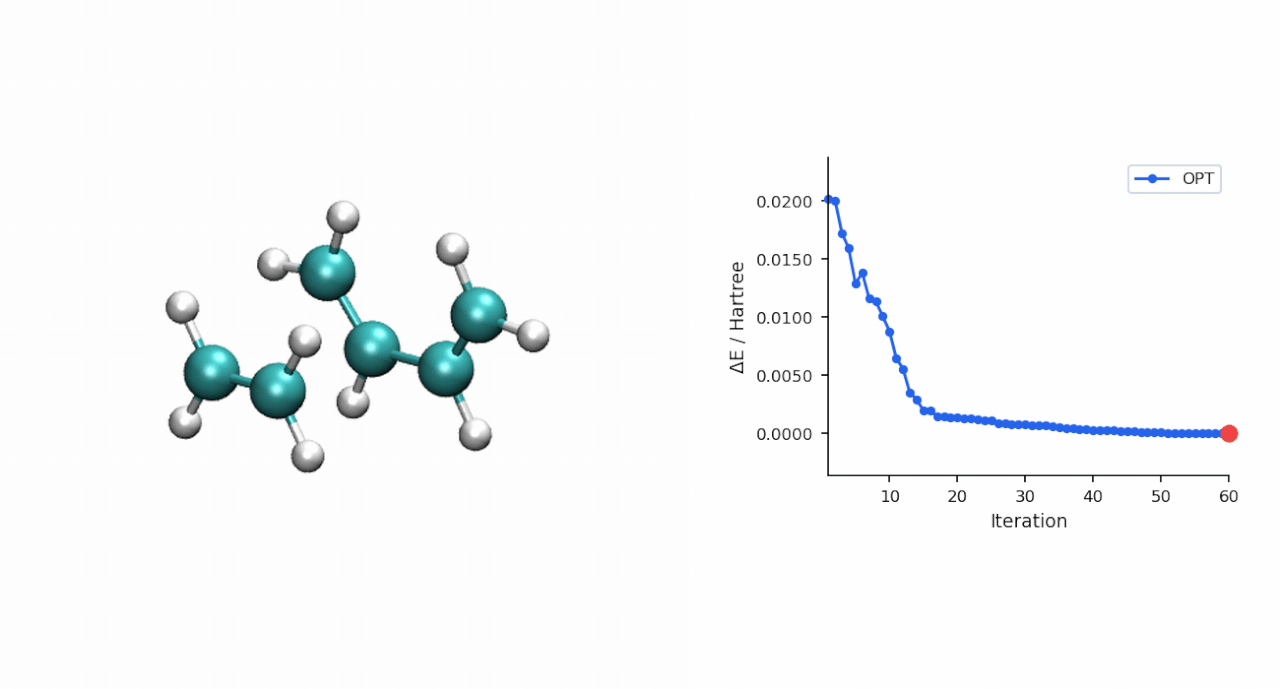
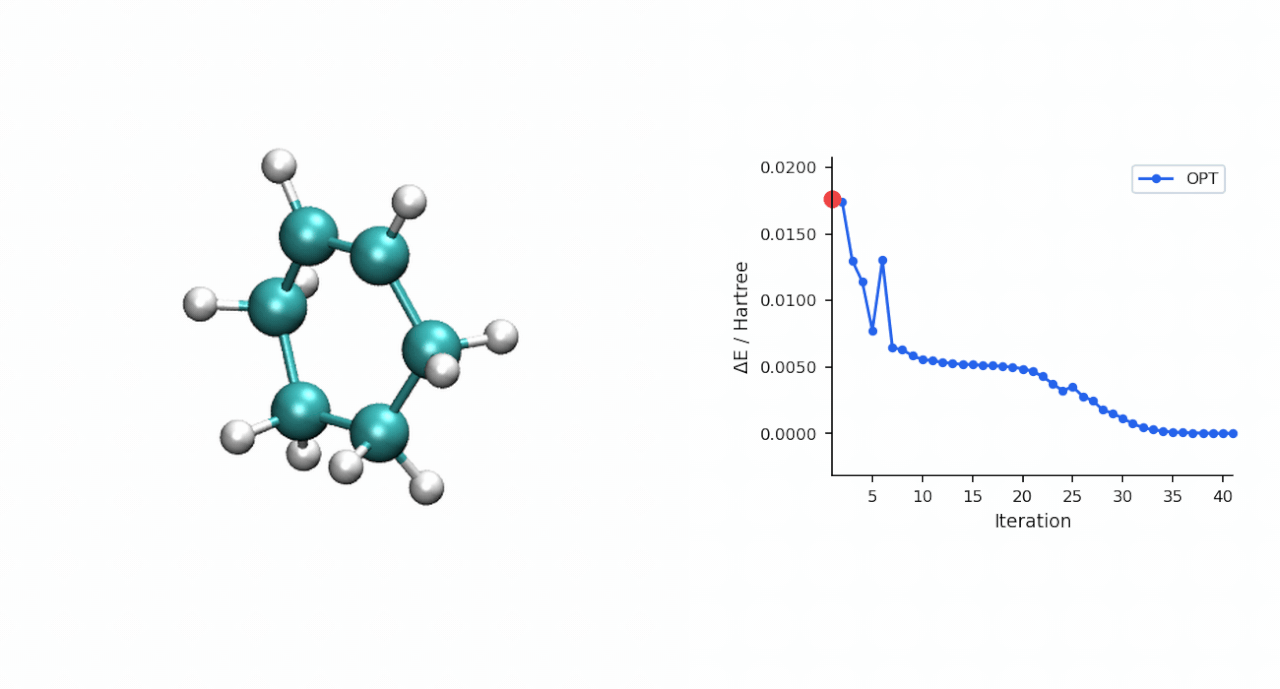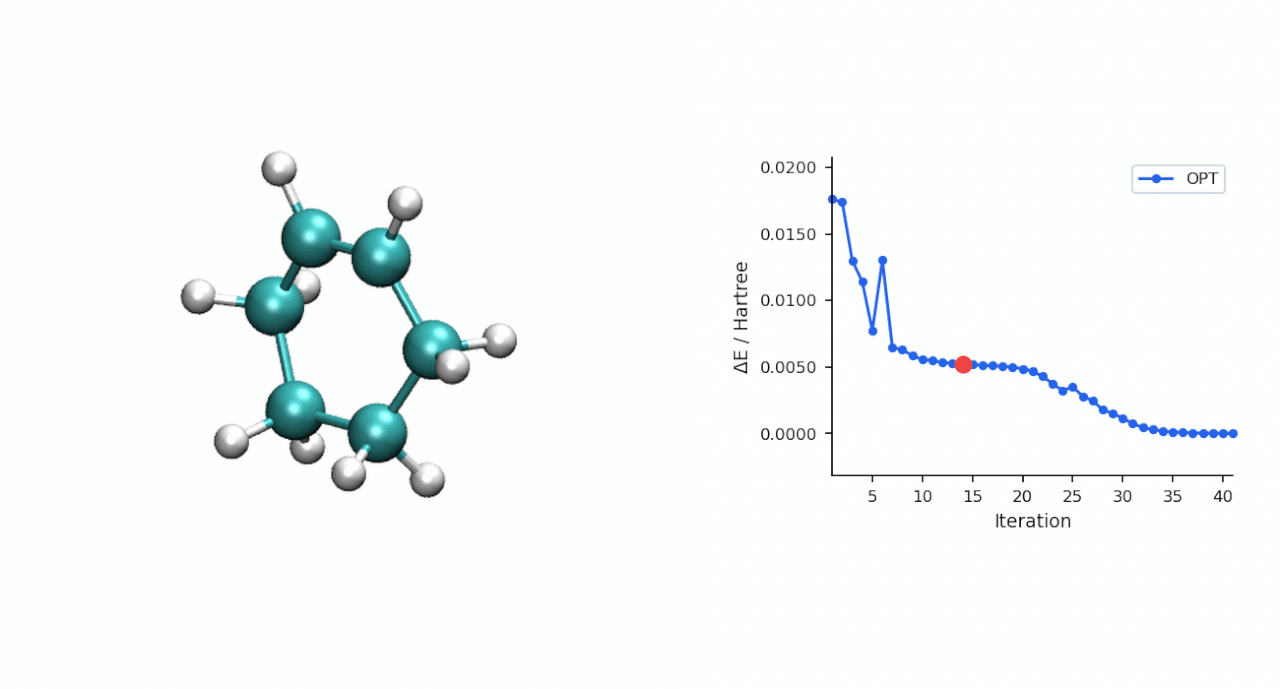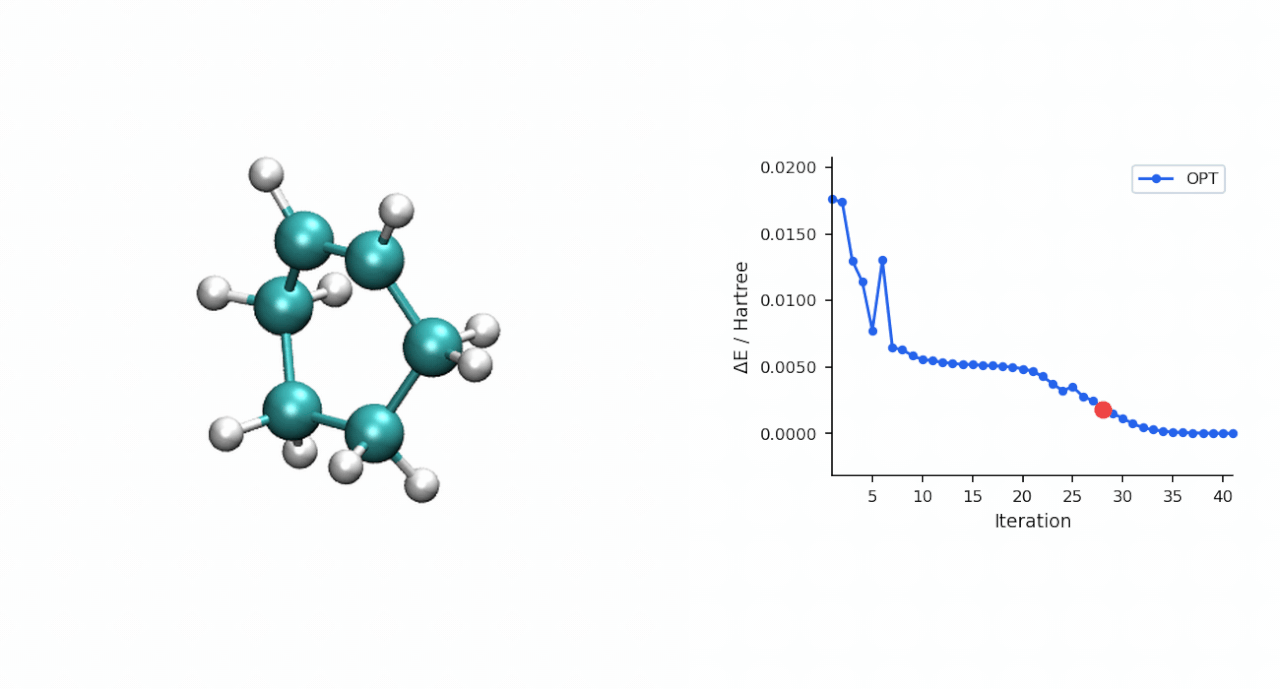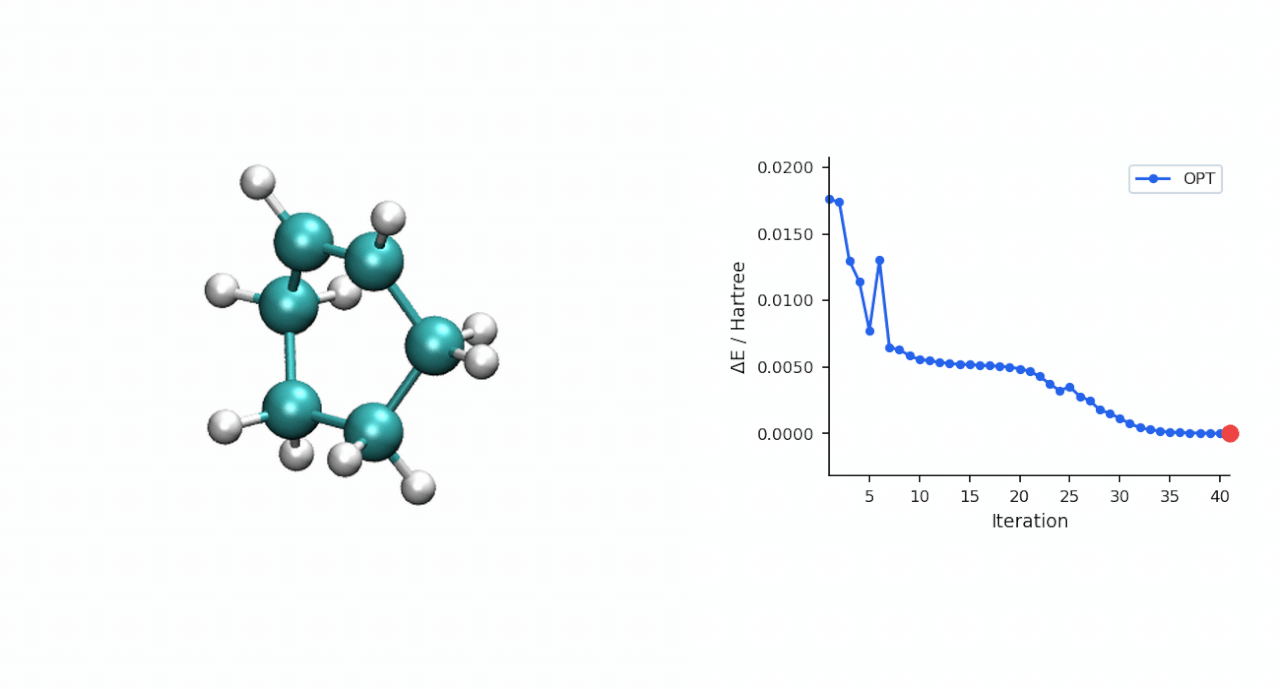
ΔE‡= E(TS) − E(R) = (−234.493874 − (−234.525473)) Eh = 19.8 kcal/mol
| 键 | TS 实测值 | 预期物理量 |
| 形成键 C0–C10 | 2.222 Å | ~2.2 Å (同步) |
| 形成键 C3–C11 | 2.222 Å | 与上严格相等 → 严格 C2v 同步 |
| 丁二烯中心 C1–C2 | 1.402 Å | 1.34 (反) ↔ 1.50 (产) 之间,介于双/单 |
| 乙烯残余 C10–C11 | 1.377 Å | 1.33 (反) ↔ 1.54 (产) 之间,部分单键 |
| C0–C1, C2–C3 | 1.372 Å (对称) | 类似中部 |
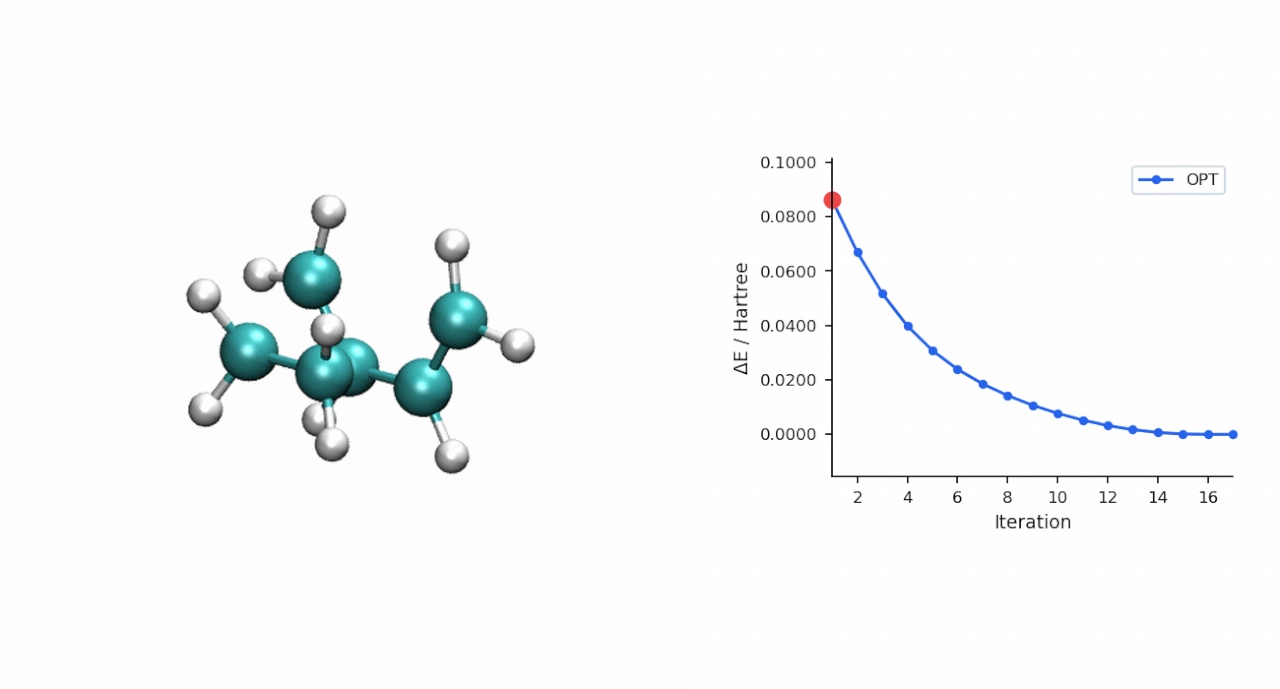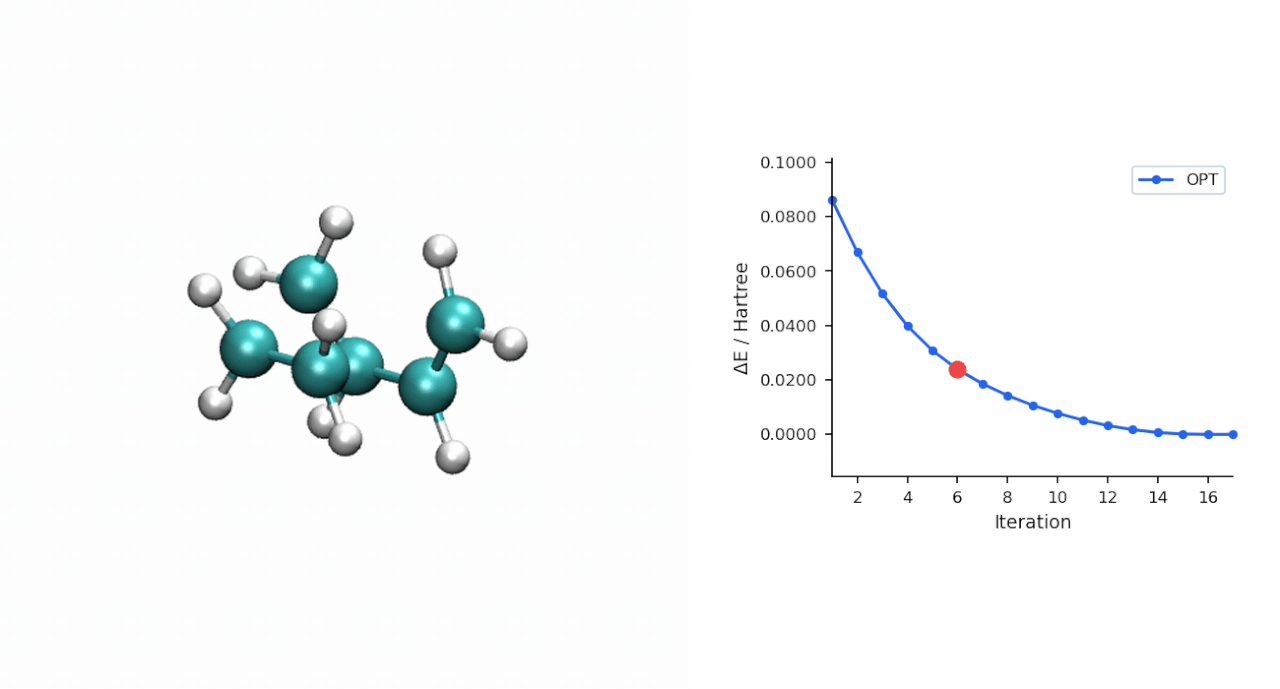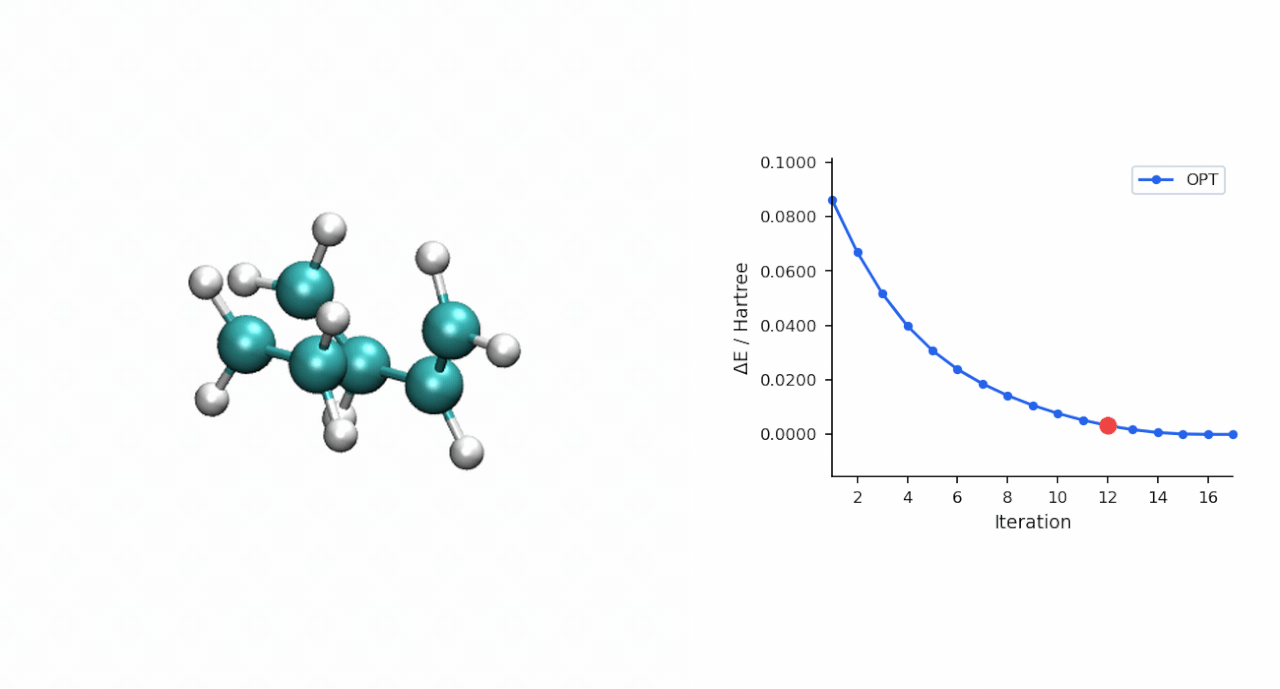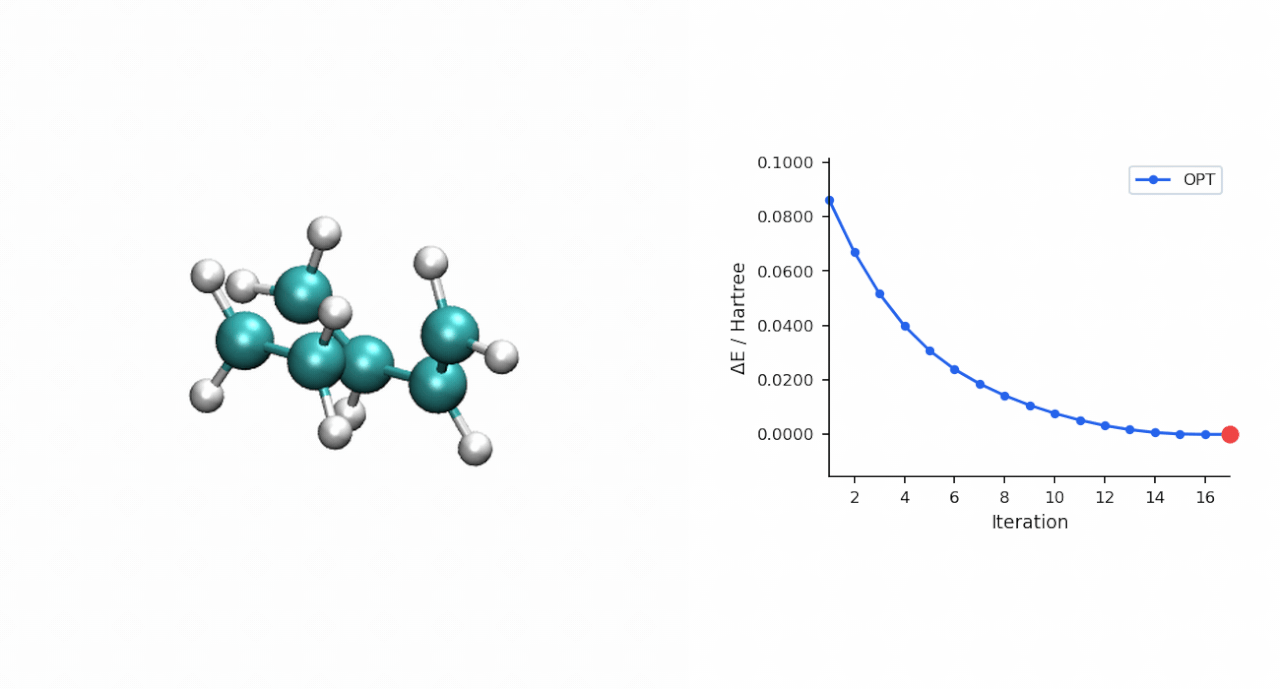
TS 初猜的经验:D-A TS 比较"温和",P-RFO 对初猜要求不高,只要形成键距离落在 ~2.0–2.5 Å 范围且对称,基本一次就能收敛。对于更复杂的过渡态(迁移反应、电环化、多步级联),如果手搓初猜不容易,可以先用 #ts(method=neb,refine=cineb) 从反应物和产物两端跑 CI-NEB(10–15 个镜像,max_iter=500),取得到的 HEI(最高能量镜像)作为 P-RFO 初猜——MAPLE 的 NEB 会自动把 HEI 写到 *_hei.xyz,可以直接接力。
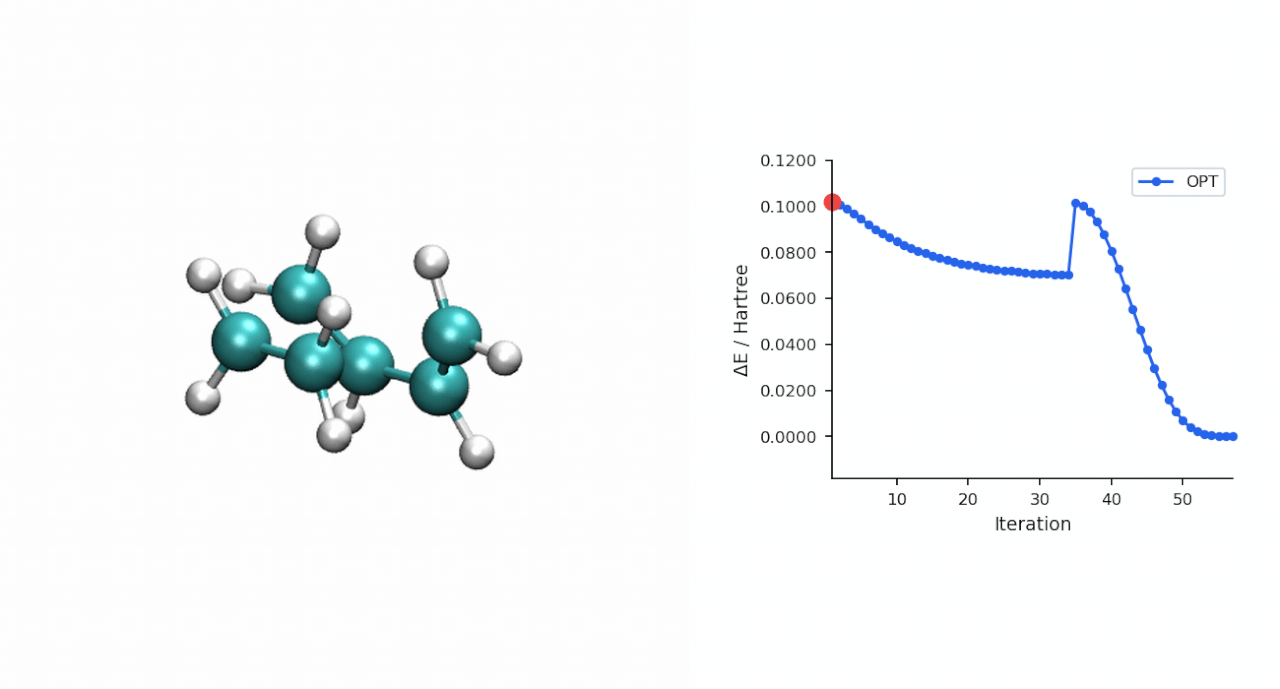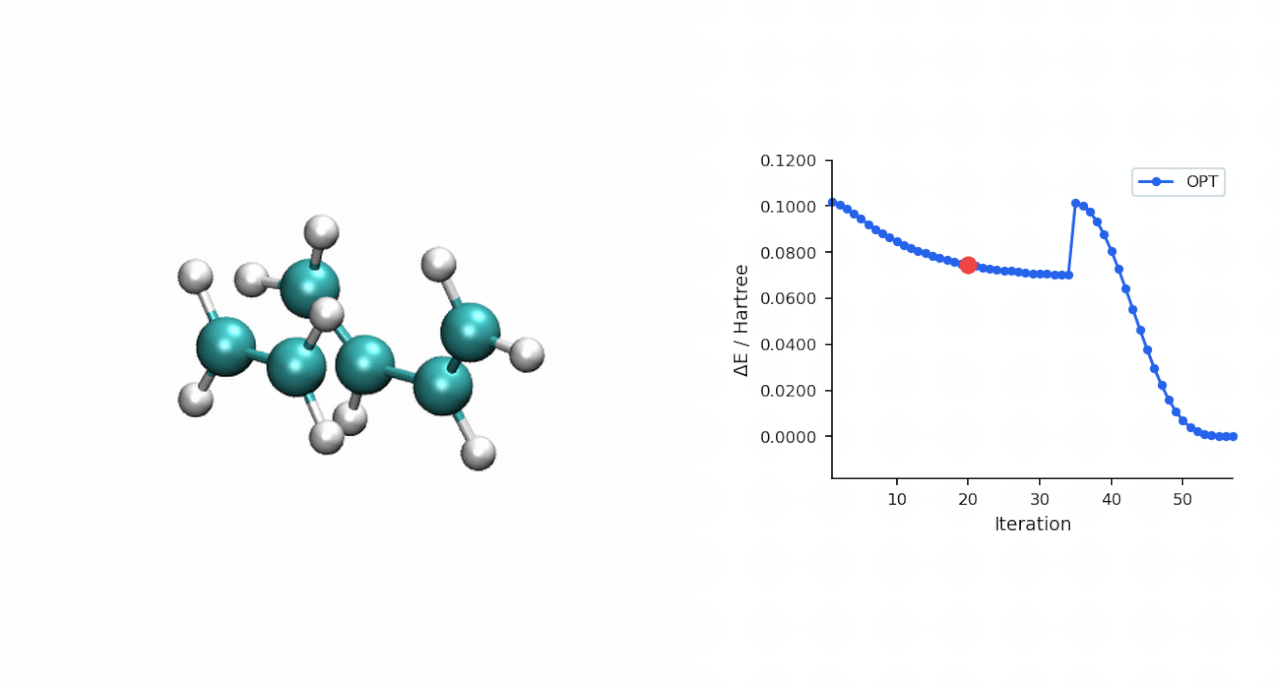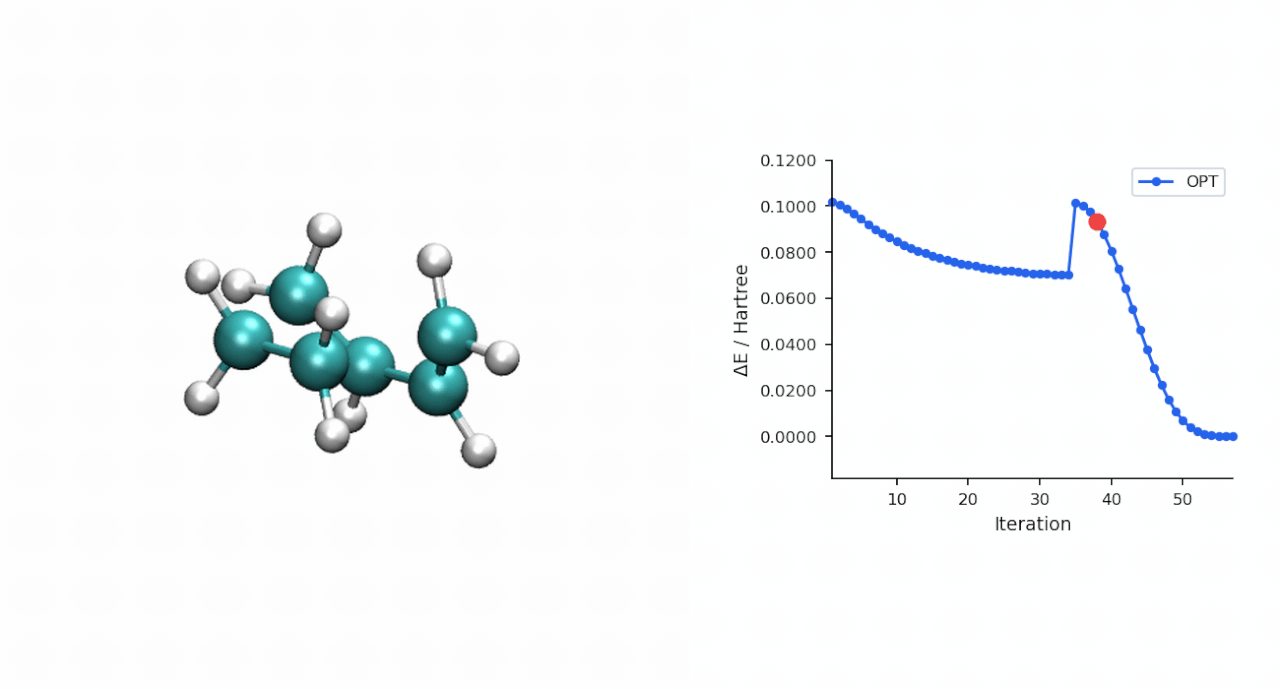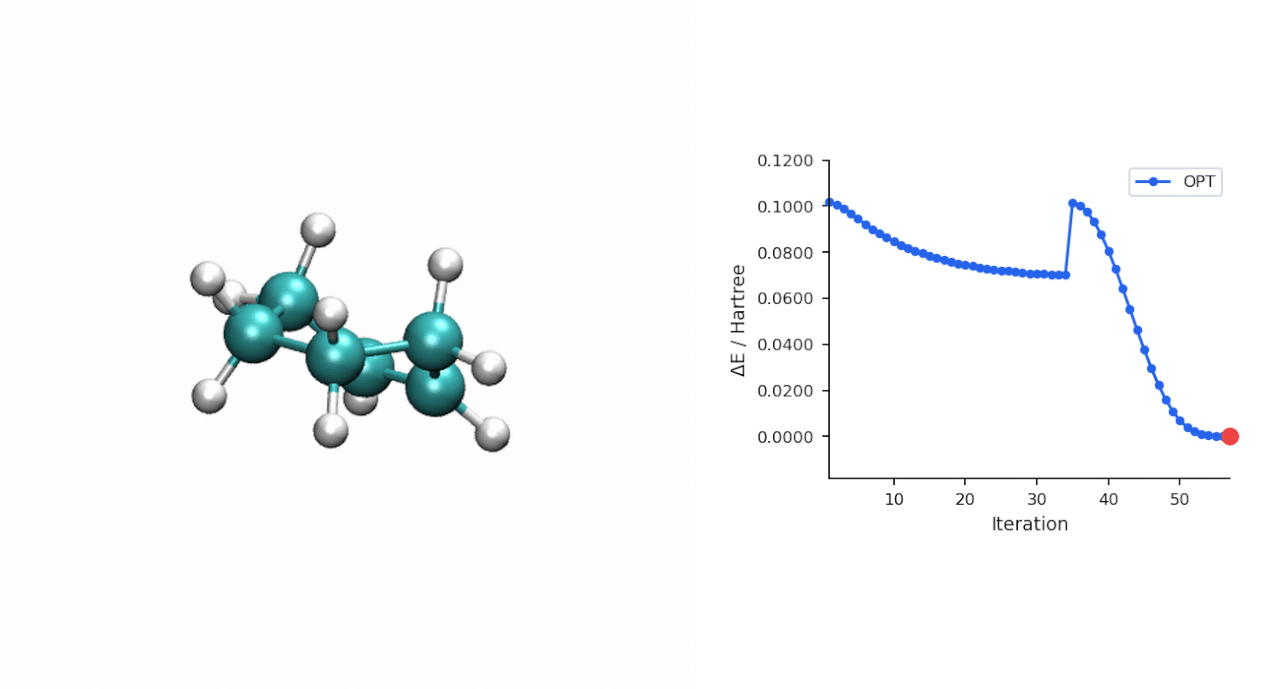
| IRC 端点 | 端点能量 (Eh) | 端点再 OPT 后 (Eh) | 关键距离 (Å) | 拓扑判定 |
| 正向 → P 侧 | −234.595732 | −234.595945 | C0–C10 = 1.539, C3–C11 = 1.538 | 环己烯产物 |
| 反向 → R 侧 | −234.525539 | −234.526176 | C0–C10 = 3.531, C3–C11 = 3.515 | pre-reactive R 复合物 |
| TS(IRC step 24) | −234.494015 | — | — | 与 P-RFO 鞍点差 0.09 kcal/mol |
注意 MAPLE 的“forward / backward”与化学的“反应物侧 / 产物侧”不存在固定对应——这取决于初始虚频方向的随机相位。本例 forward 落到 P 侧、backward 落到 R 侧; 但 forward/backward 与化学反应方向没有固定对应,最终仍应以端点优化后的拓扑和能量判断。
步骤 | 实际耗时 | 输出 |
| 3.1 反应物预优化 | 18.5 s | R 优化结构、E_R = −234.525473 Eh |
3.1 产物预优化 | 13.1 s | P 优化结构、E_P = −234.595945 Eh |
3.2 P-RFO 精修 TS | 167.9 s | 同步 TS, ΔE‡ = 19.8 kcal/mol, C—C 形成 2.22 Å × 2 |
3.3 频率分析 | 21.6 s | 1 虚频 −627.43 cm⁻¹, 41 实频 |
3.4 IRC(双向) | 53.5 s | 57 步全连通,full/forward/backward 三个轨迹文件 |
3.4 IRC 端点 OPT(正向) | 10.8 s | 环己烯(−234.595945 Eh,与 P 严格一致) |
3.4 IRC 端点 OPT(反向) | 9.6 s | pre-reactive R 复合物(−234.526176 Eh) |
完整流程合计 | 295.0 s (~4.9 分钟) | R → TS → freq → IRC → 端点 OPT |
UMA: Liu R. et al. UMA: Wood B. M. et al. UMA: A Family of Universal Models for Atoms. DOI: 10.48550/arXiv.2506.23971.
牧生 发表于 2026-5-19 07:17
请帮忙解决一下,由于网络问题,我的服务器没法直连这个文件。我在win下载了,传到MAPLE目录,仍然提示找 ...
牧生 发表于 2026-5-19 07:59
我是用的pip install -e . 但是/MAPLE/maple/function/calculator/model 里面是空的。。现在把ani1xnr.pt ...
DwyaneWan 发表于 2026-5-19 01:17
非常棒的工作。请问是否支持周期性,无限延展得有机聚合物得变胞优化呢?
牧生 发表于 2026-5-19 12:07
请能不能把command_control.py提及的这些都传到网盘啊。
SUPPORTED_MODELS = {
"ani2x",
mizu-bai 发表于 2026-5-19 22:31
Congrats! 非常好工作,爱来自重庆。
说起来我从更早的 sander 中实现 ML/MM 的项目就关注了,从 MAPLE ...
Huschein 发表于 2026-5-20 16:32
MAPLE短时间内不会有MLMM的计划 因为引入MM部分会是很大的工作量而且会让程序本身很臃肿 MLMM建议还是到A ...
ljh123 发表于 2026-5-21 21:48
请问这项工作目前或者未来是否计划支持微调的大模型呢?
AxiEJohn 发表于 2026-5-22 06:43
谢谢提问,这是一个好问题!这让我们想得更多。
就目前来说,如果是基于MAPLE支持的模型系列微调的,您 ...
ljh123 发表于 2026-5-22 11:28
谢谢!我相信如果能支持微调模型的话,应用范围将会大大提升
Marucs 发表于 2026-6-12 00:47
请问MAPLE支持多个任务同时进行么?比如说一个任务同时跑OPT 和 FREQ
Feel free to criticize!
BeMgCaSrBa 发表于 2026-7-2 16:57
不支持win啊,太可惜了
| 欢迎光临 计算化学公社 (http://bbs.keinsci.com/) | Powered by Discuz! X3.3 |