Q-Chembio-llg 发表于 2023-1-16 22:00
如果不想用高斯的话可以考虑VASP
kuroha 发表于 2023-1-16 16:06
有点小白,不知道如何用高斯来做,自学的课程没见过处理分子筛这种大体系,请问有什么教程或者论文能拜读 ...
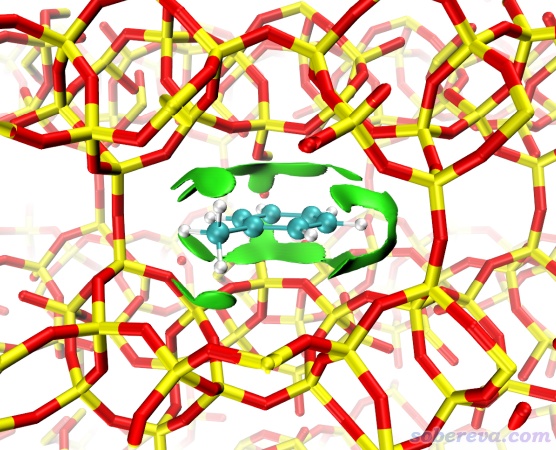
sobereva 发表于 2023-1-17 05:21
好好记住Gaussian怎么拼
用簇模型,从分子筛中挖出一块,对边界饱和,优化时冻结边界原子,用Gaussian计 ...
| 欢迎光临 计算化学公社 (http://bbs.keinsci.com/) | Powered by Discuz! X3.3 |